
Recrystallization Mechanisms of Aluminum and Aluminum Oxide Interfaces through Reactive Simulations
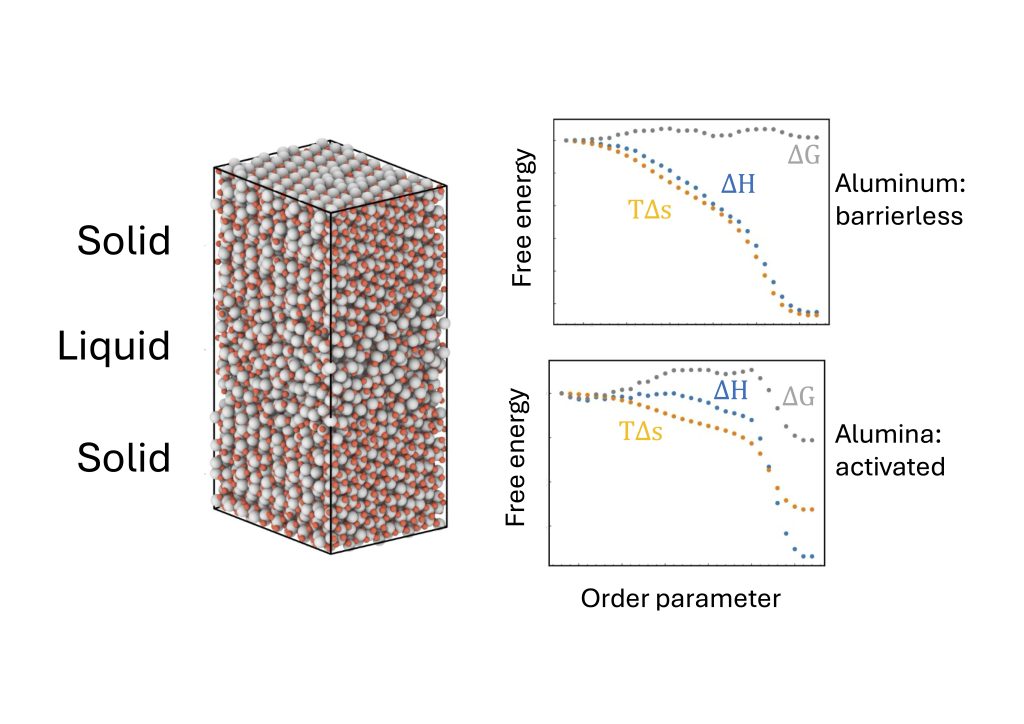
This study used reactive molecular dynamics (ReaxFF) and unsupervised clustering to analyse how aluminium and alumina crystallise from the melt.
Aluminium atoms crystallise rapidly via a barrierless process, while alumina grows more slowly through a sequential mechanism: oxygen atoms incorporate first, facing significant free energy barriers, followed by aluminium atoms. Per-atom Gibbs free energies and bond-orientational order parameters revealed that atomic charge, especially for oxygen, strongly influences alumina crystallisation.
These findings explain the contrasting crystallisation kinetics of metals and metal oxides and provide microscopic insight for materials design.
Authors: Hao Zhao & Fernando Bresme