

- This event has passed.
TYC Distinguished Speaker Symposium: Modelling Surfaces & Catalysis
30 March 2022 @ 2:00 pm – 5:00 pm
Watch the Symposium here
Professor Dr Joachim Sauer – Humboldt Universität zu Berlin
Next Generation Quantum Chemistry of Water in Acidic Zeolites
Water plays a ubiquitous role in the synthesis, post-synthesis treatment and reactivity of zeolite catalysts. We consider the local structure of bridging OH groups (b-OH) and their interaction with water molecules for Al-O(H)-Si sites in different zeolite frameworks and at different locations.
A review of past work is followed by next generation studies which go beyond previous work in several respects:
(1) Structure optimizations are performed at the MP2 level using our hybrid MP2(cluster model):DFT-D(periodic) method.
(2) Special attention is paid to sites with internal H-bonds across rings of corner-sharing TO4 tetrahedra to a Si-O-Si acceptor site (Fig., left).
(3) In addition to the H-bond approach of H2O to the b-OH site (Fig., right), we consider Lewis attack to the AlO4 tetrahedron in anti-position to the b-OH site (Fig., middle).
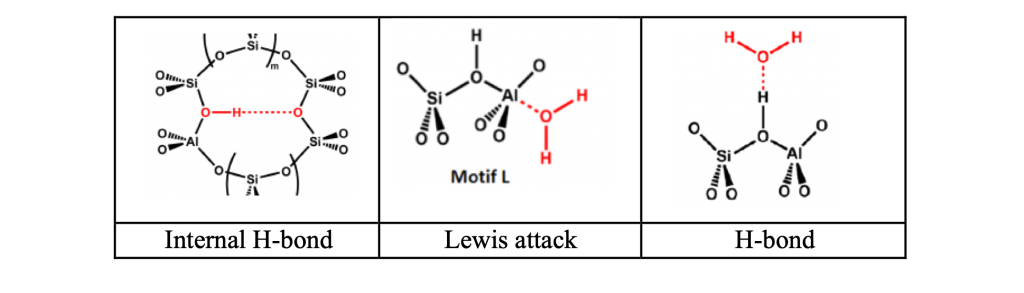
The MP2-quality results for OH vibrational frequencies and 1H-NMR chemical shifts of different types of bridging OH groups and their interaction with one and two water molecules are compared with experimental spectroscopic results and heats of adsorption.
The conclusions are also relevant for adsorption of alcohols at b-OH sites.
Professor Adrian Mulholland – University of Bristol
Multiscale modelling of biocatalysts for enzyme design, evolution and engineering
Simulations are revealing biomolecular mechanisms relevant to function, and are contributing to catalyst and inhibitor design. Simulations can be used as computational ‘assays’ of biological activity, e.g. to predict drug resistance or effects of mutation. Combined quantum mechanics/molecular mechanics (QM/MM) methods allow modelling of reactions in proteins: they can identify mechanisms of reaction (e.g. for targeted covalent inhibitors such as ibrutinib, and for the SARS-CoV-2 main protease) determinants of catalytic activity and predict the activity of bacterial enzymes against antibiotics.
Dynamical-nonequilibrium molecular dynamics (D-NEMD) simulations show coupling between allosteric sites and the active site in beta-lactamase enzymes; the pathways identified contain positions that differ between clinically relevant variants, indicating that allosteric effects modulate the spectrum of activity of these antibiotic resistance enzymes. The D-NEMD approach can effectively combine cloud-based and other HPC resources.
Increasingly, simulations are contributing to the engineering of natural enzymes and de novo biocatalysts. Simulations are also contributing to the emerging evidence that activation heat capacity is an important factor in enzyme evolution and thermoadaptation. Directed evolution of a designed Kemp eliminase unexpectedly introduced curvature into the temperature dependence of reaction, showing the emergence of an activation heat capacity. Simulations identify the dynamical networks involved, which may provide useful targets for mutation and directed evolution experiments.
Virtual reality offers new ways interact with simulations, and new ways to collaborate. Interactive MD simulation in virtual reality (iMD-VR) allows manipulation of biological macromolecules, going beyond mere visualization to allow e.g. fully flexible docking of drugs into protein targets. The COVID-19 pandemic has highlighted the need for effective tools for virtual collaboration. Groups of researchers can work together, using iMD-VR for molecular problems such as catalyst and structure-based drug design. Using the cloud, researchers in different physical locations can work together in the same virtual molecular environment. Simulations, including iMD-VR, with collaborative sharing of models and data, have been brought together to design peptide inhibitors of the SARS-CoV-2 main protease.
References
‘Evolution of dynamical networks enhances catalysis in a designer enzyme HA Bunzel, JL Anderson, D Hilvert, VL Arcus, MW van der Kamp & AJ Mulholland Nature Chemistry 13, 1017-1022 (2021)
‘Designing better enzymes: Insights from directed evolution’ HA Bunzel, JLR Anderson & AJ Mulholland Current Opinion in Structural Biology 67, 212-218 (2021)
‘Dynamical nonequilibrium molecular dynamics reveals the structural basis for allostery and signal propagation in biomolecular systems ASF Oliveira, G Ciccotti, S Haider, AJ Mulholland The European Physical Journal B 94, 1-12 (2021)
‘Discovery of SARS-CoV-2 M pro peptide inhibitors from modelling substrate and ligand binding H. Chan et al. Chemical Science 12, 13686-13703 (2021)
Biography: Adrian Mulholland is a Professor of Chemistry, University of Bristol, UK. Following his first degree at Bristol, he worked in a wine merchant and for ICI Pharmaceuticals before doctoral studies with Graham Richards (Oxford) and postdoctoral work with Martin Karplus (Harvard). His research focuses on mechanisms of enzyme catalysis, biomolecular dynamics and function. He develops and applies biomolecular simulation methods to problems in antimicrobial resistance, drug metabolism, biocatalysis and enzyme design and evolution. He has published over 200 papers, attracting over 10,000 citations. He was awarded the 2020 John Meurig Thomas Medal ‘for outstanding and innovative work in catalytic science’.
Dr Thomas Keal – Science and Technology Facilities Council (STFC)
Recent developments in QM/MM modelling with ChemShell
ChemShell is a scriptable computational chemistry environment with an emphasis on multiscale simulation of complex systems using combined quantum mechanical and molecular mechanical (QM/MM) methods. The QM/MM approach is well suited to studying catalysis in both biomolecular systems and materials, where the reactive region can be treated at the QM level and the environment with classical methods. QM/MM is particularly useful when coupled with serial crystallography experiments, as is highlighted by a case study of the mechanism of nitrite reduction in a copper nitrite reductase enzyme [1]. Recent work in QM/MM modelling of materials chemistry will also be discussed, as well as the redevelopment of ChemShell as an open source, python-based package, which offers a modern platform for multiscale modelling with an emphasis on high performance computing platforms [2].
References:
[1] K. Sen, M.A. Hough, R.W. Strange, C. Yong and T.W. Keal, J. Phys. Chem. B, 125, 9102 (2021).
[2] Y. Lu, M.R. Farrow, P. Fayon, A.J. Logsdail, A.A. Sokol, C.R.A. Catlow, P. Sherwood and T.W. Keal, J. Chem. Theory Comput., 15, 1317 (2019).
Biography: Thomas Keal is a Principal Scientist in the Computational Chemistry Group at STFC Daresbury Laboratory, with responsibility for QM/MM methods development. He completed his PhD in 2005 in the group of David Tozer at Durham University, focussing on the development of new exchange-correlation functionals for density functional theory. He then moved to a postdoctoral position in the group of Walter Thiel in Mülheim an der Ruhr, Germany, working on methods for excited state optimisation and dynamics of biomolecules. He joined Paul Sherwood’s group at Daresbury in 2008 to continue work on methods development in the ChemShell software package, and now leads the team developing ChemShell. His research interests are in QM/MM methodology and its application to problems in biochemistry and materials chemistry.
Dr Edina Rosta – University College London
Dynamics, function and mechanism of phosphate processing enzymes
Phosphate catalytic enzymes are essential and ubiquitous to all forms of life. While structures of these proteins are typically readily available, prediction and design of their function and activity is a key current challenge. Here we review free energy calculation methods and applications for prototype examples including HIV-1 RNase H [1]. Our work highlights the important role of coupled proton transfer steps in the catalytic mechanism using the finite-temperature string method. This allows us to use multiple collective variables that govern the reaction path. Identification of these collective variables in complex processes presents a major problem. We offer promising AI-driven algorithms to help identify essential reaction coordinates in biomolecular processes [2].
Edina Rosta1, Department of Physics and Astronomy, University College London, London, WC1E 6BT, e.rosta@ucl.ac.uk
References
[1] S. Dürr, O. Bohuszewicz, R. Suardiaz, P. G. Jambrina, C. Peter, Y. Shao, and E. Rosta, ACS Catalysis, 10.1021/acscatal.1c01493, 2021
[2] M. Badaoui, P. J. Buigues, D. Berta, G. M. Mandana, H. Gu, T. Földes, C. J. Dickson, V. Hornak, M. Kato, C. Molteni, S. Parsons, and E. Rosta, J. Chem. Theory Comput. 10.1021/acs.jctc.1c00924, 2022
Biography: Dr. Edina Rosta is an Associate Professor in Computational Materials Modelling at UCL. After completing her PhD at USC in the group of Arieh Warshel (2013 Chemistry Nobel Prize Laureate), she joined the Hummer lab as a Postdoctoral Research Fellow at the NIDDK, NIH. She took up a lecturer position at KCL Chemistry in 2012. In 2020 she joined UCL Physics. Current research in her group focuses on atomistic molecular modeling, including hybrid quantum mechanics/molecular mechanics (QM/MM) simulations. To quantitatively and accurately assess how enzymes achieve their extraordinary efficiency and specificity in performing chemical reactions, she develops modern enhanced sampling methods including novel algorithms to calculate molecular kinetics from biased molecular simulations using the theoretical framework of kinetic networks. Applications of her work focus on the most prominent chemical reactions of living organisms: phosphate transfer and cleavage. She studies the key functional roles of Mg2+ cofactors in phosphate catalytic reactions.
Organised by:
Professor Sir Richard Catlow
tyc-administrator@ucl.ac.uk