
Global Optimization of Molybdenum Subnanoclusters on Graphene: A Consistent Approach toward Catalytic Applications
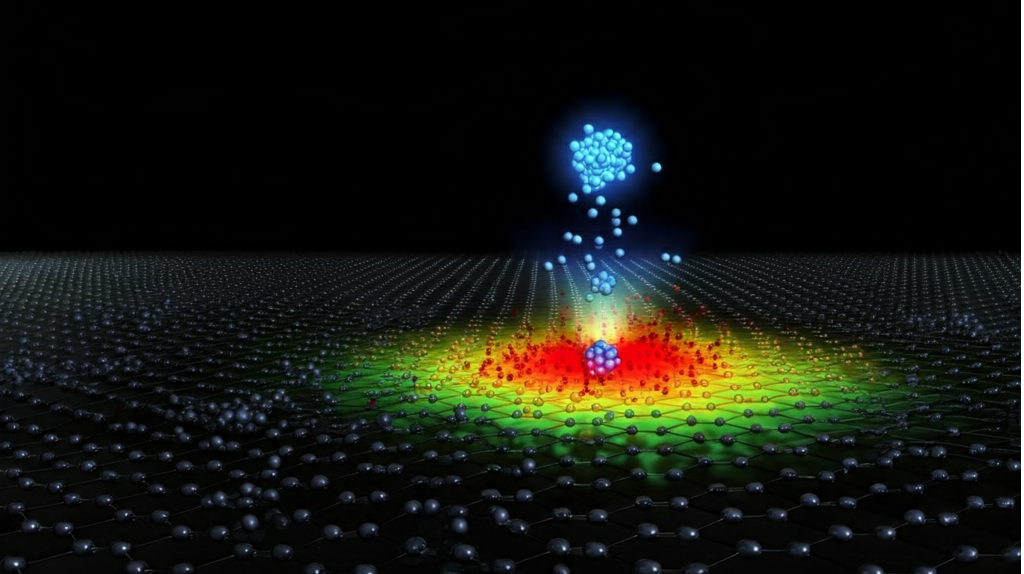
The design of novel subnanometer cluster (SNC) catalysts with enhanced catalytic properties necessitates precise control over the clusters’ size, shape, and deposition onto surfaces. However, the inherent complexity of the adsorption process hinders a comprehensive understanding of the structure-reactivity relationships crucial for rational catalyst design. Conventional computational methods often rely on limited structural sampling, neglecting the complex energy landscape and potentially leading to inaccurate predictions. Furthermore, these methods frequently overlook the experimental deposition process, which can significantly influence the prevalence of specific adsorption geometries, including metastable configurations.
This study presents a novel approach employing global optimisation techniques, specifically the particle swarm optimisation (PSO) method, integrated with ab-initio calculations. This approach simulates the entire experimental process, from predicting SNC structures in the beam and on the surface to evaluating their reactivity. As an example, we studied the deposition of a molybdenum SNC consisting of 6 atoms onto a free-standing graphene surface, and its catalytic activity towards CO dissociation. While illustrative, the calculations provide valuable insights into the complex energy landscape of Mo SNCs on graphene, highlighting their catalytic potential and the importance of comprehensive configurational sampling. This study establishes a robust framework for theoretically driven rational catalyst design.
Authors: Yao Wei, Alejandro Santana-Bonilla, Lev Kantorovich