
Mechanochemistry of phosphate esters confined between sliding iron surfaces
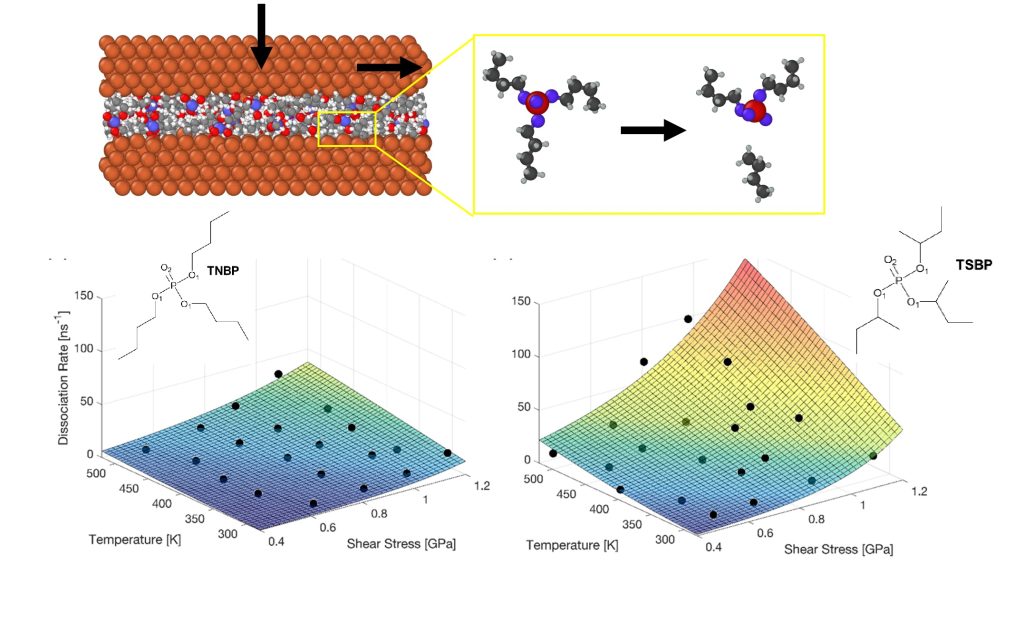
The molecular structure of lubricant additives controls not only their adsorption and dissociation behaviour at the nanoscale, but also their ability to reduce friction and wear at the macroscale. Here, we show using nonequilibrium molecular dynamics (NEMD) simulations with a reactive force field (ReaxFF) that tri(s-butyl)phosphate dissociates much faster than tri(n-butyl)phosphate when heated and compressed between sliding iron surfaces. We perform NEMD simulations over a wide range of temperatures and pressures. Using a modified Arrhenius equation that accounts for the reduction in the activation energy due to the applied shear stress (the Bell model), we show that the main difference between the trialkyl phosphates is mainly due to the pre-exponential factor, rather than the activation energy or the activation volume. This observation is consistent with macroscale tribometer experiments with similar lubricant additives. This study represents a crucial step towards the virtual screening of lubricant additives with different substituents to optimise tribological performance.
Authors: Carlos Ayestarán Latorre, Joseph E. Remias, Joshua D. Moore, Hugh A. Spikes, Daniele Dini & James P. Ewen
Featured in: Communications Chemistry volume 4, Article number: 178 (2021)