
Understanding the photochemistry of a crystalline push–pull norbornadiene photoswitch
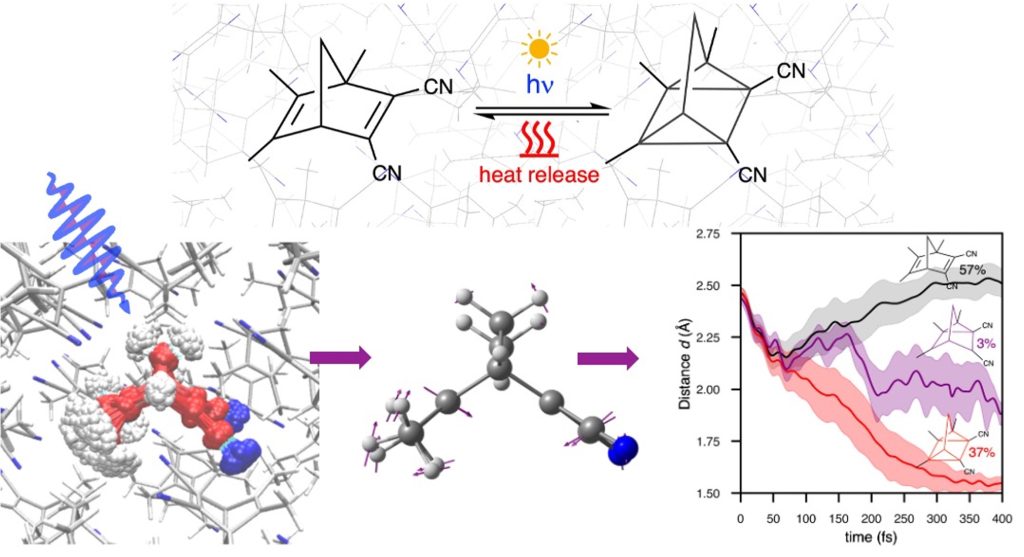
Capturing solar energy in chemical bonds and releasing it on demand is a promising route towards a sustainable energy future. So-called Molecular Solar Thermal (MOST) materials achieve exactly this: functional molecules based on molecular photoswitches absorb sunlight, twist into a high-energy “charged” metastable isomer, and later relax back, releasing the stored energy as heat on demand. Solid, crystalline MOST materials are especially attractive because they pack a high density of photoactive molecules into a small volume and are easy to integrate into devices — but how the surrounding crystal lattice steers the underlying photoreaction remains largely a black box. In this study, we present a novel state-of-the-art multiscale quantum-mechanical simulation and non-adiabatic molecular dynamics protocol to resolve, atomistically, what happens inside a crystal of a “push–pull” norbornadiene photoswitch immediately after it absorbs light. Our calculations show that the crystal packing leaves just enough free volume for the molecule to undergo the large structural rearrangement needed to reach its high-energy quadricyclane form via an ultrafast [2+2] photocycloaddition, with a remarkably high quantum yield (37%) and an energy density (0.36 MJ kg⁻¹) above the practical threshold for solar-fuel applications. Beyond this specific material, the work introduces an openly available, transferable computational framework for probing ultrafast photochemistry directly in molecular crystals, paving the way for the rational, atomistic design of the next generation of solid-state materials for harvesting and storing solar energy.
Authors: Federico J.Hernández, Jordan M.Cox,Jingbai Li, StevenLopez, Rachel Crespo-Otero